blast
下载blast
1 | wget -c ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-2.10.0+-x64-linux.tar.gz |
构建数据库
1 | makeblastdb -in genome.fasta -dbtype nucl -parse_seqids -out ./index |
-in:构建数据库所用的序列文件。
-dbtype:数据库类型。构建的数据库是核苷酸数据库时,dbtype设置为nucl,数据库是氨基酸数据库时,dbtype设置为prot。以核苷酸数据库为例
-out:数据库名称。建议以建库基因组的名字来命名,做到见名知意。
以石松的线粒体序列为例:

建完库后会出现以下文件:
序列比对
构建好数据库就可进行序列比对。序列比对的工具共有5种,大家可以根据自己序列比对的类型进行选择。以blastn为例。
blastn:将核苷酸序列比对至核苷酸数据库。
blastp:将氨基酸序列比对至氨基酸数据库。
blastx:将核苷酸序列比对至氨基酸数据库。
tblastn:将氨基酸序列比对至核苷酸数据库。比对时,将输入的氨基酸序列与数据库中核苷酸序列翻译后的氨基酸序列逐一比对。
tblastx:将核苷酸序列比对至核苷酸数据库。与blastn的区别是比对时,输入的核苷酸序列与数据库中的核苷酸序列都先翻译为氨基酸序列,而后再进行逐一比对。
以blastn为例,进行序列比对。
1 | ## 将核苷酸序列比对至核苷酸数据库 |
-query:进行检索的序列。
-db:使用的数据库。
-evalue:设置输出结果中的e-value阈值。e-value低于1e-5就可认为序列具有较高的同源性。
-outfmt:输出文件的格式,一般设置为6。
-num_threads:线程数。
-out:输出文件。做到见名知意。
例:
之后会出现一个结果文件

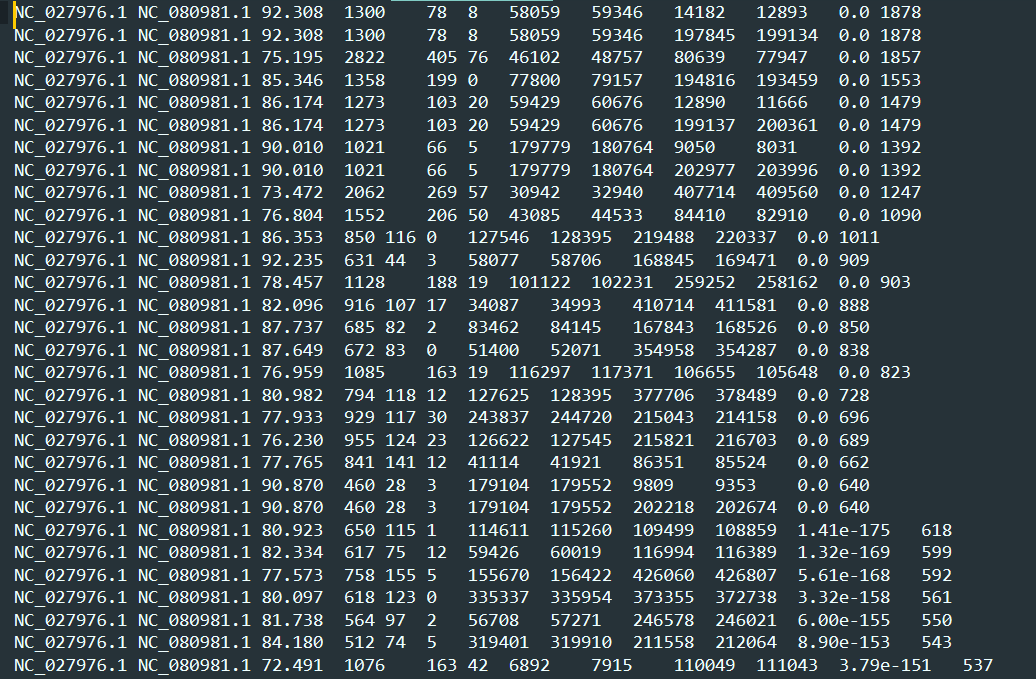
第1列:输入序列的名称。
第2列:比对到的目标序列名称。
第3列:序列相似度。
第4列:比对的有效长度。
第5列:错配数。
第6列:gap数。
第7-8列:输入序列比对上的起始和终止位置。
第9-10列:比对到目标序列的起始和终止位置。
第11列:e-value。
第12列:比对得分。
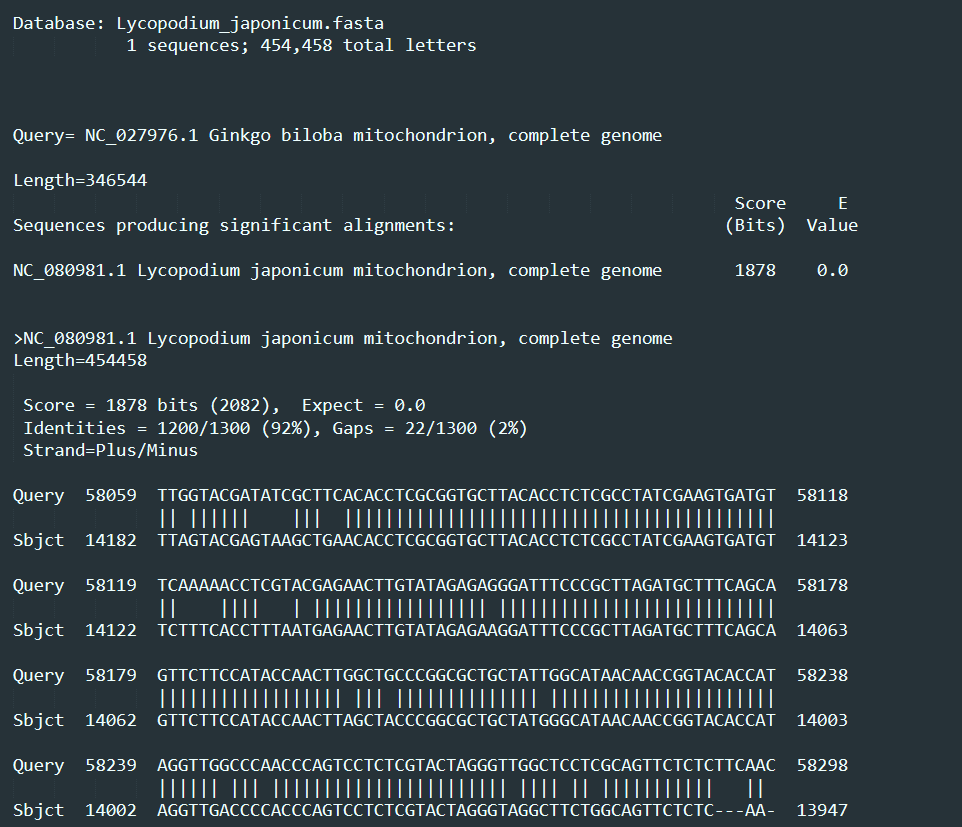
如果想要知道序列每个碱基的比对情况,删去outfmt参数即可。
1 | blastn -query input.fa -db ./index -evalue 1e-6 -num_threads 6 -out out_file |