
PhyloSuite使用教程
打开PhyloSuite
- 解压压缩包,找到PhyloSuite.exe并双击打开
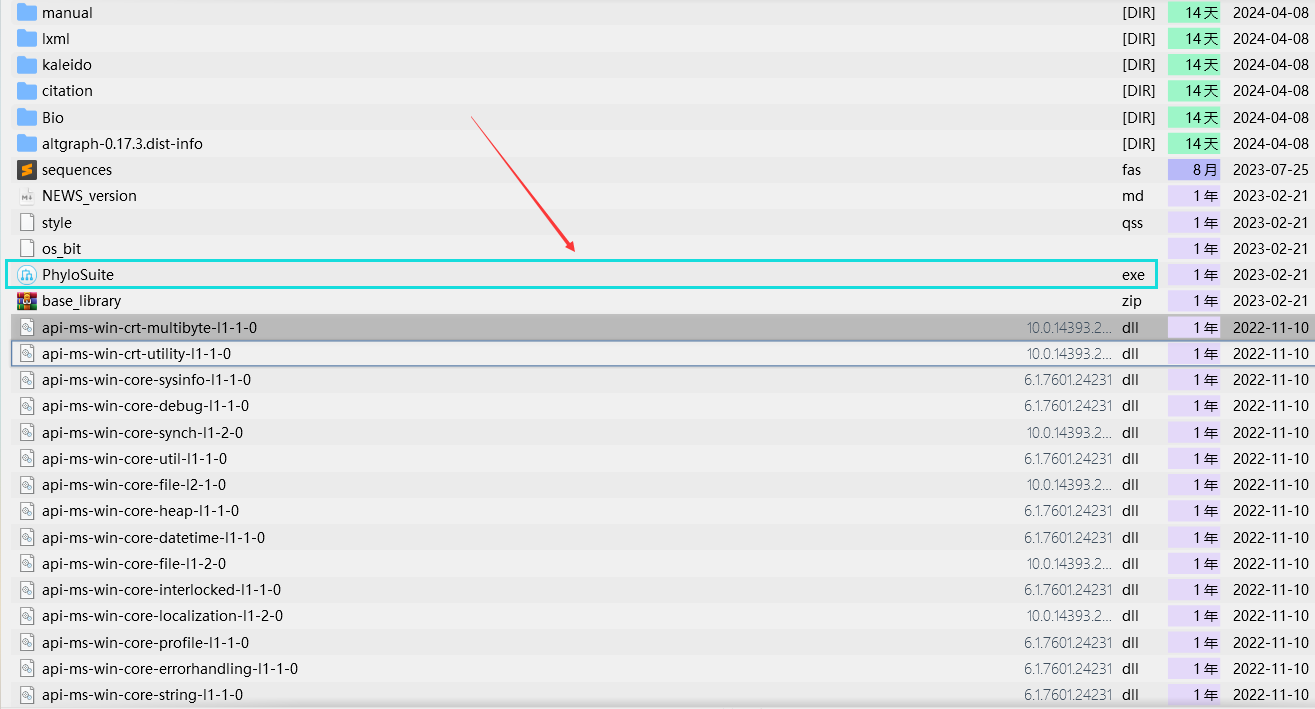
PhyloSuite的使用
使用PhyloSuite进行系统发育分析需要用到线粒体或叶绿体的.gb文件
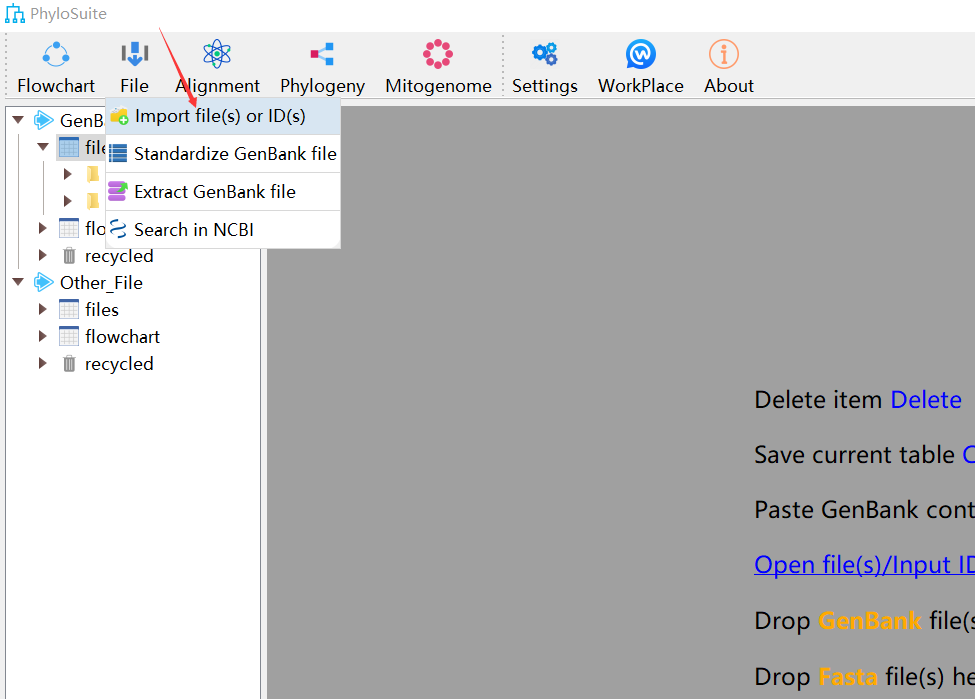
- 按照下图所示步骤进行操作导入gb文件,在此,我们以本地选择文件(a)为例。选择Arabidopsis thaliana(NC_037304)和Cucurbita pepo(NC_014050 )两个物种。
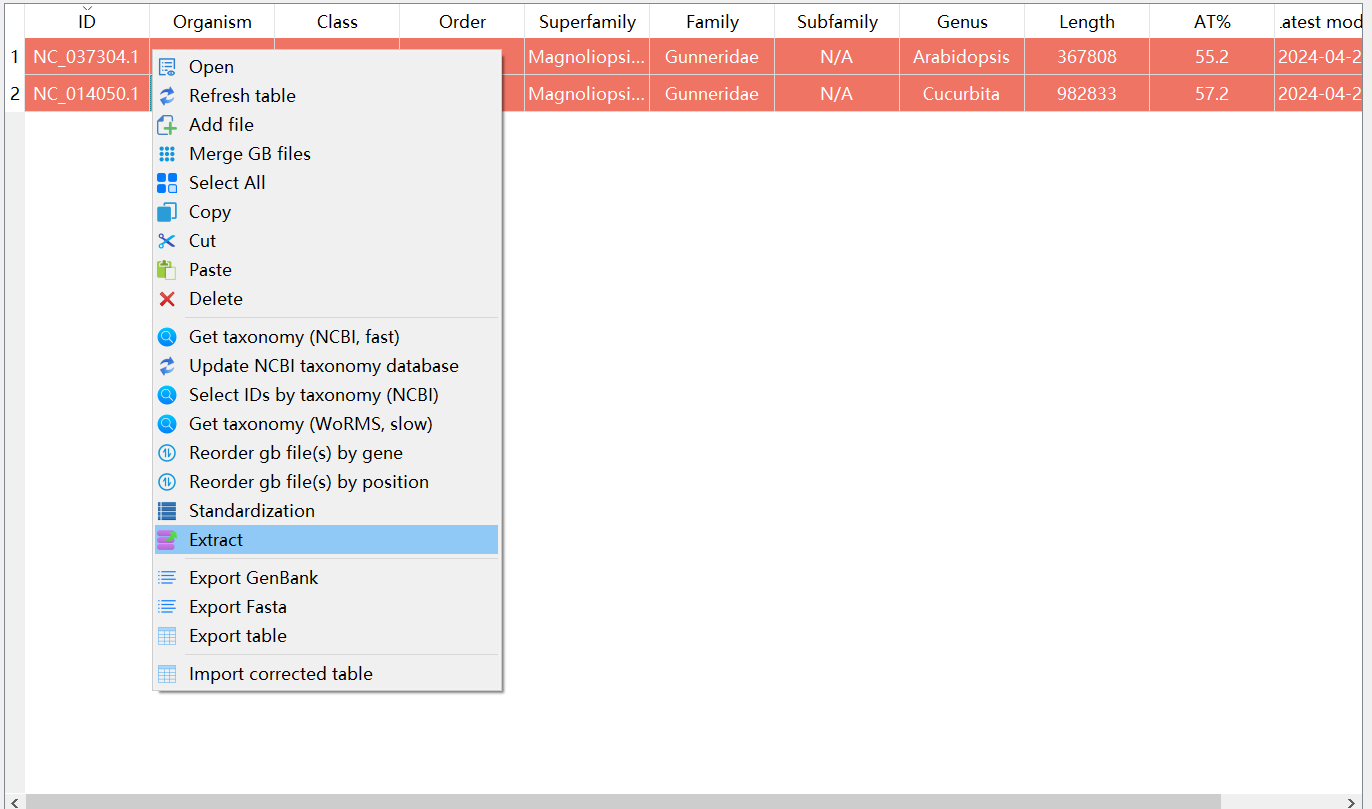
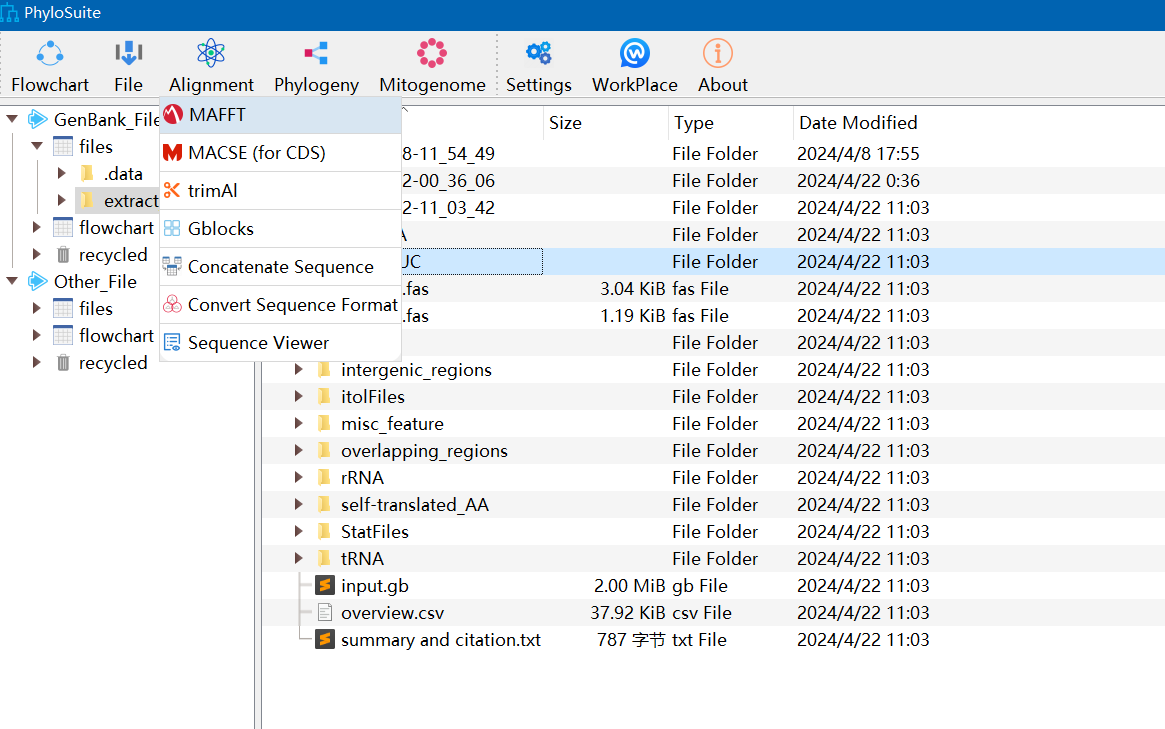
- Extract导入的gb文件:在导入成功后,我们对导入的文件进行Extract,如下图所示。
- 如果你分析的是线粒体就选择线粒体,叶绿体同上。这里我们以线粒体为例。选择好后,其余参数一般保持不变,然后点击start即可。
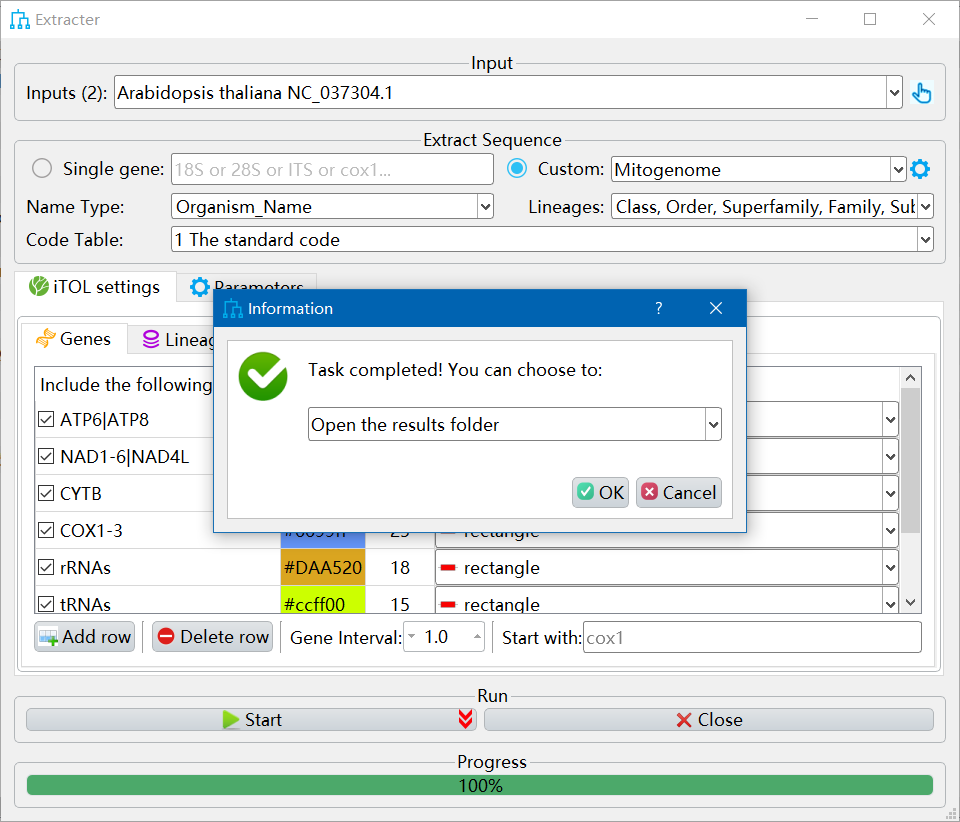
- 运行完成后,会出现如下图所示的界面,直接点击ok会在文件管理器中打开,点击cancel则会直接关闭窗口
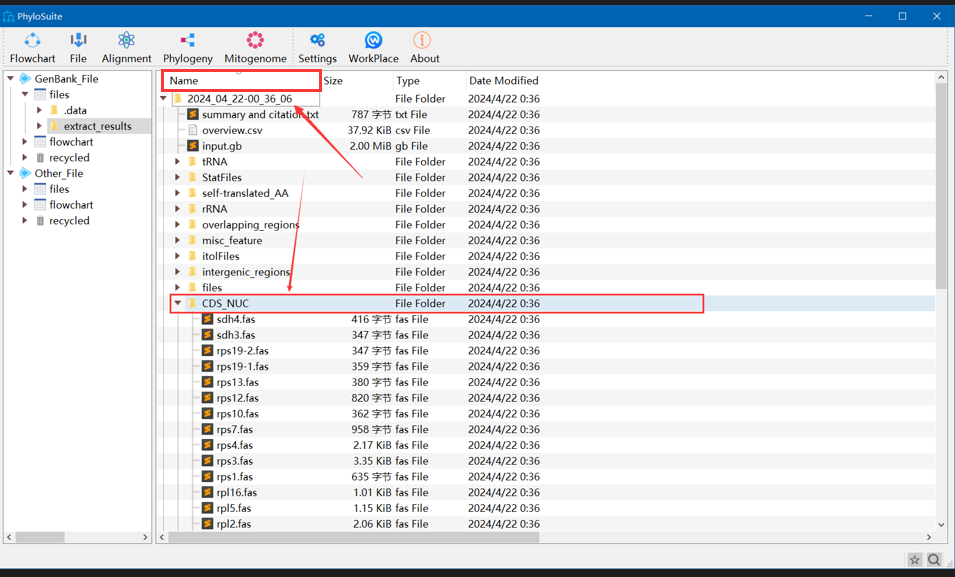
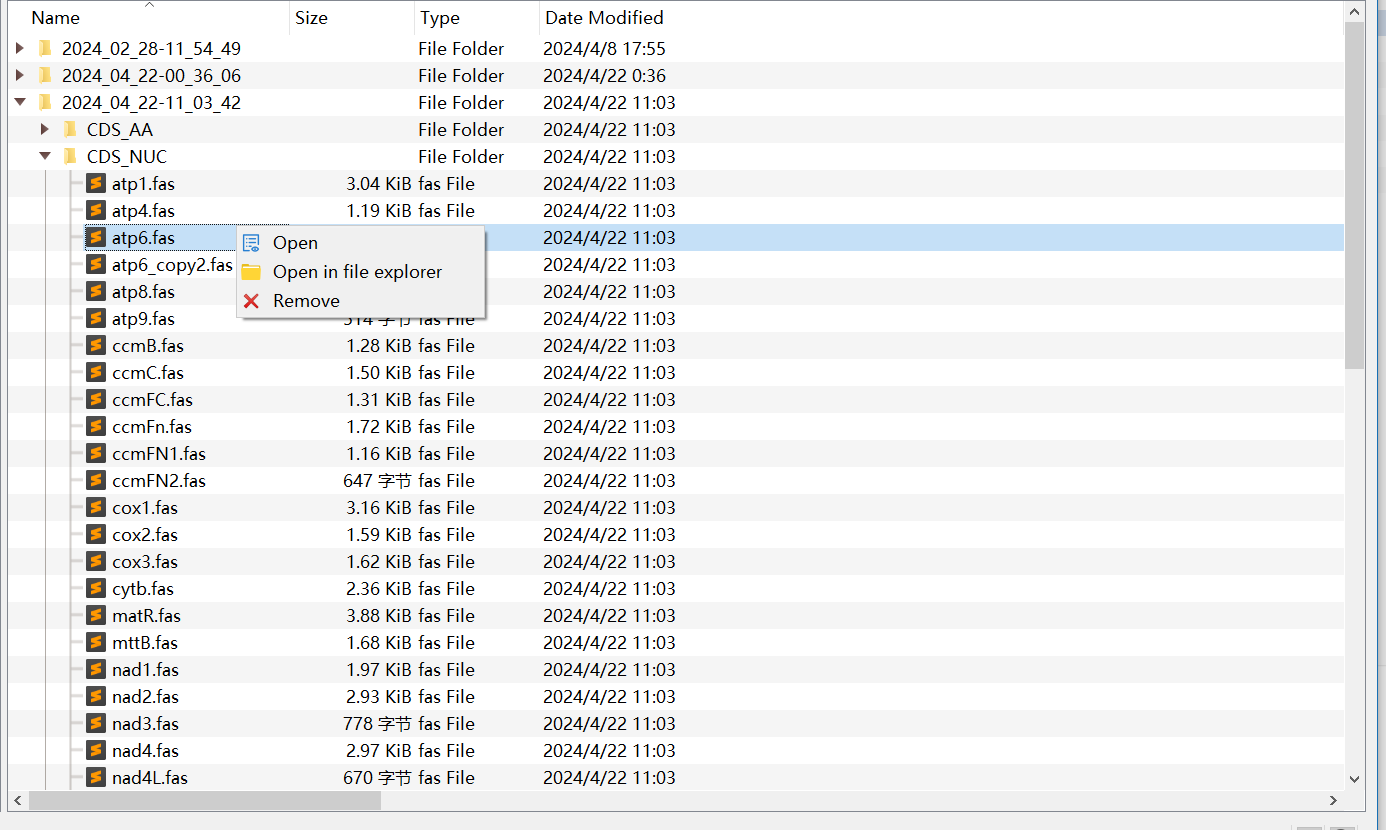
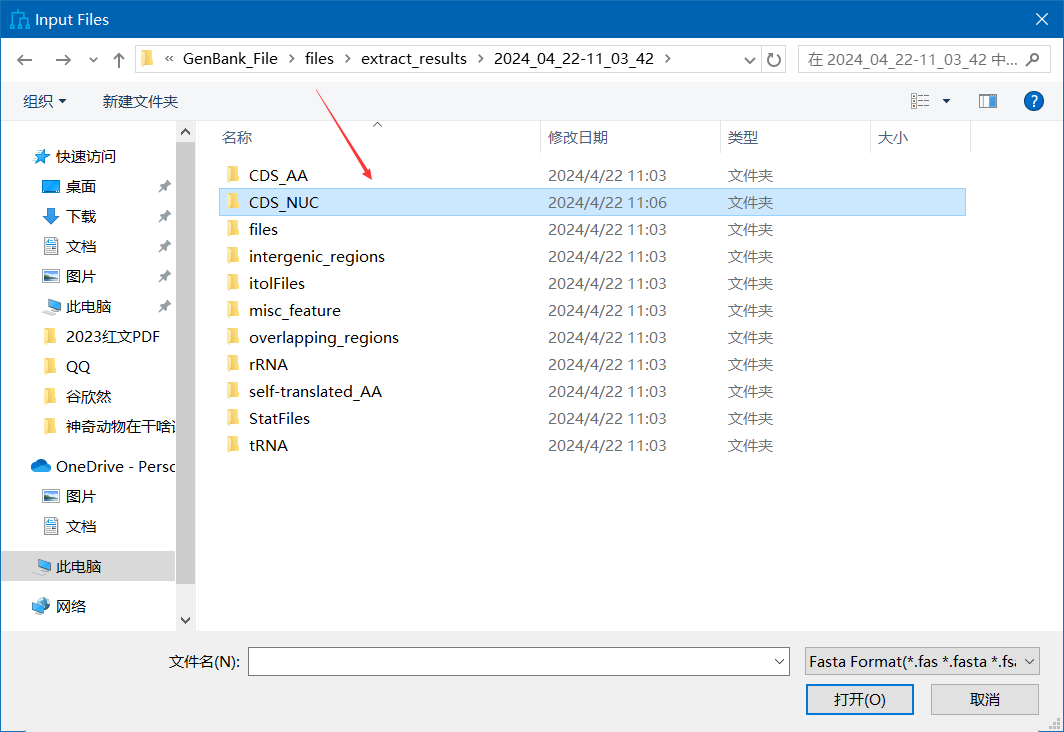
- 在程序的主界面,会出现如下内容,我们点击CDS_NUC。建议可以点击上面的name将文件排序一下。
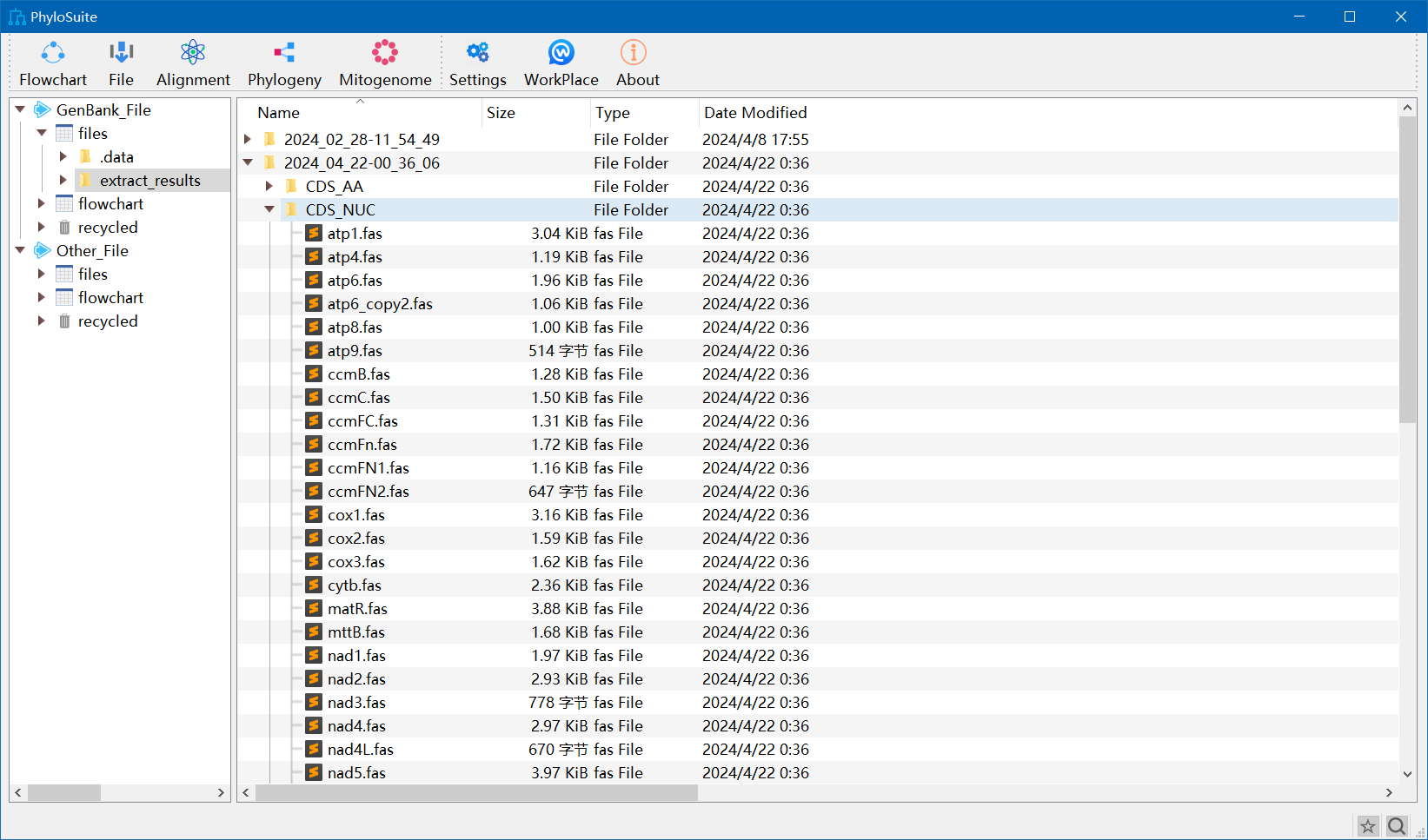
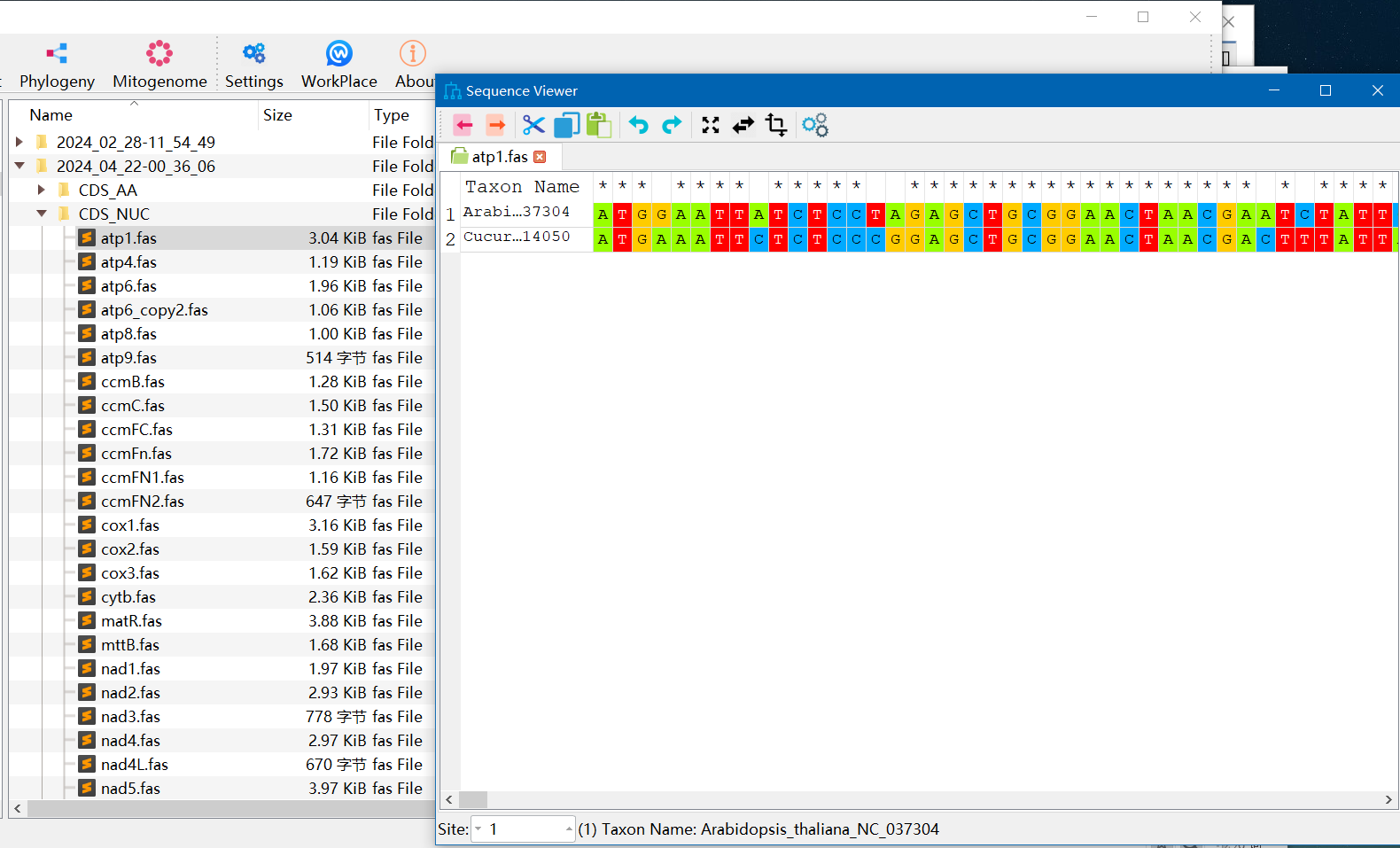
筛选共有基因:接下来,我们在点开CDS_NUC之后,要进行共有基因的筛选。共有基因越多,构建的系统发育树越准确假设你选择了20个物种进行系统发育分析,那么共有基因就是这20个物种都包含的基因。以atp1.fas为例,双击atp1.fas,如果在下图右边所示的窗口中有20行数据(也就是20个物种),那么atp1就可以选。如果没有20个,那这个基因就不可以选,即使有19个物种也不行。如果不是共有基因,可以右键直接删除。
当然,具体情况要具体分析,比如由于注释不统一的问题,相同基因名称可能会不一样,另外也要考虑基因别称的因素。此外,如果在很多基因中,都是只有某一个物种缺少这些基因,那么可以考虑删除这个物种。
在此,为了方便,我以两个物种的基因别称 基因名 orf25 atp4 orfB atp8 orfX mttB NAD nad ATP atp COX cox ccmFN ccmFn ccmFN ccmFn ccmFC ccmFc atp1- atp1 atp6- atp6 atp8- atp8 ccmC- ccmC cob- cob cox2- cox2 nad4L- nad4L rps19- rps19 rps7- rps7 sdh3- sdh3 mat-R matR coxI cox1 coxIII cox3 tatC mttB
- MAFFT序列比对
筛选完共有基因后,使用mafft进行序列比对。
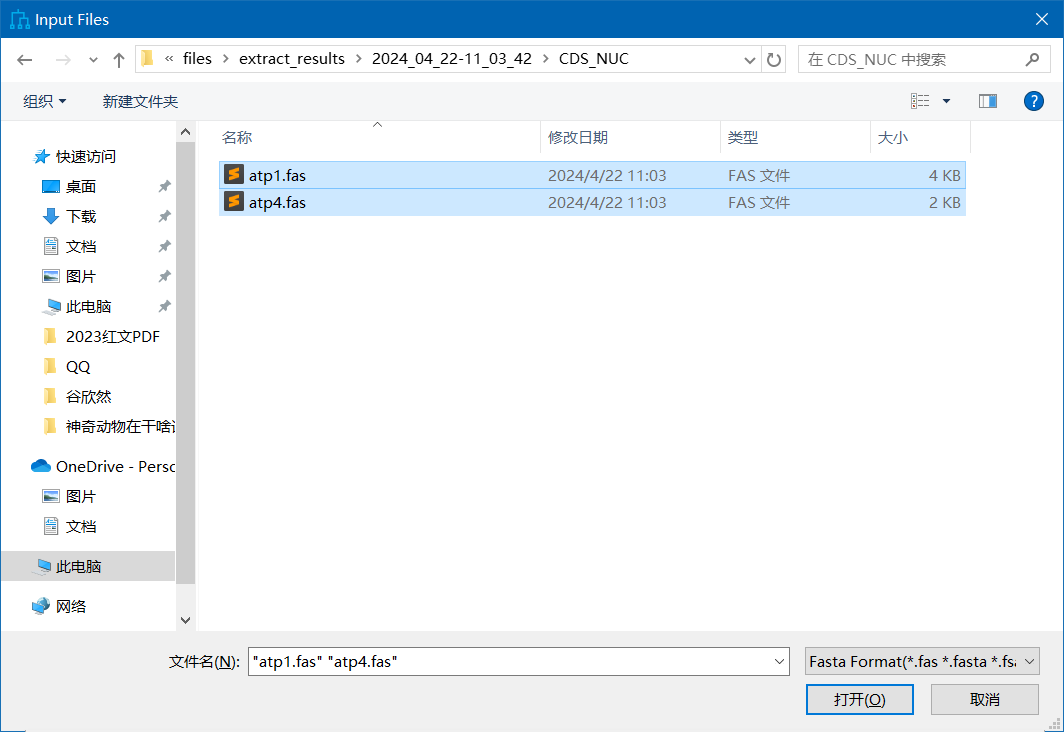
点击文件夹,选择之前筛选的共有基因文件夹,
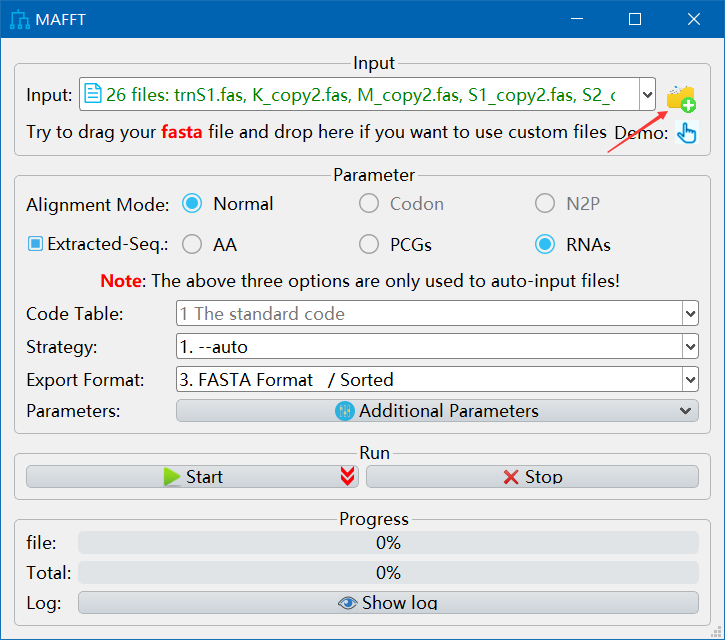
ctrl+A选择全部,点击打开
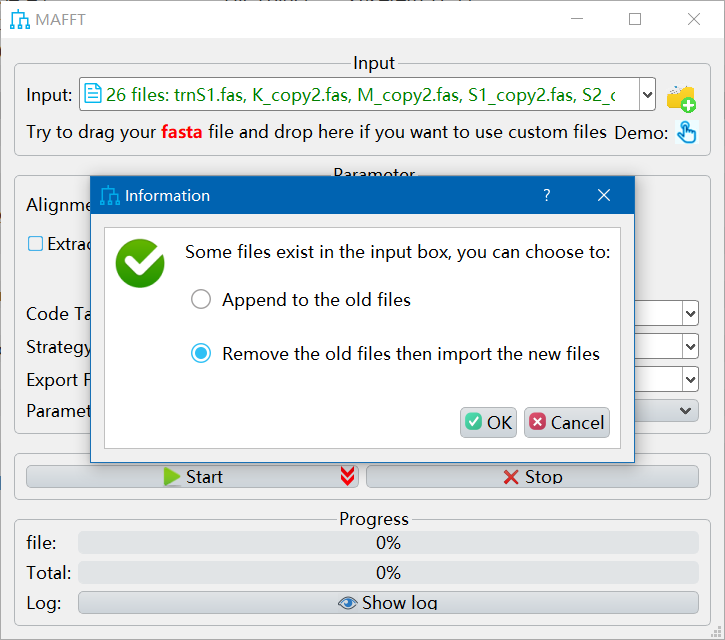
进行如下图所示的操作,点击ok
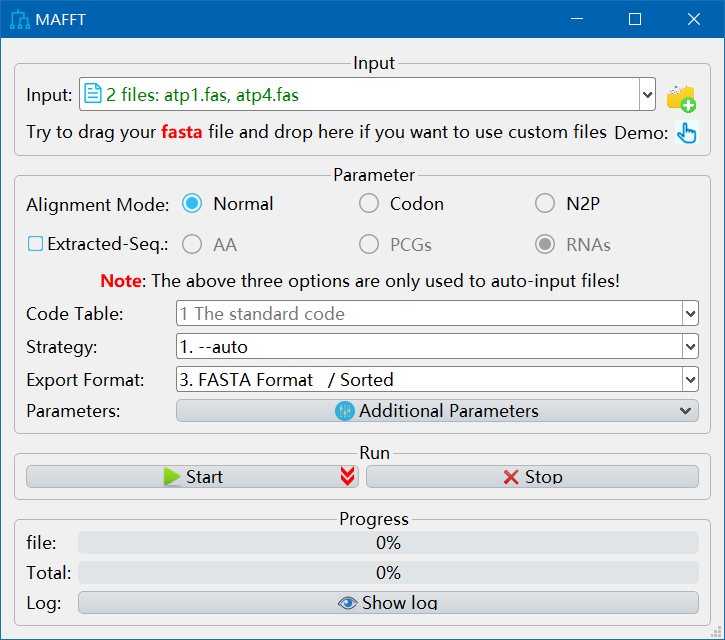
点击start,注意不要有中文路径
- 连接序列
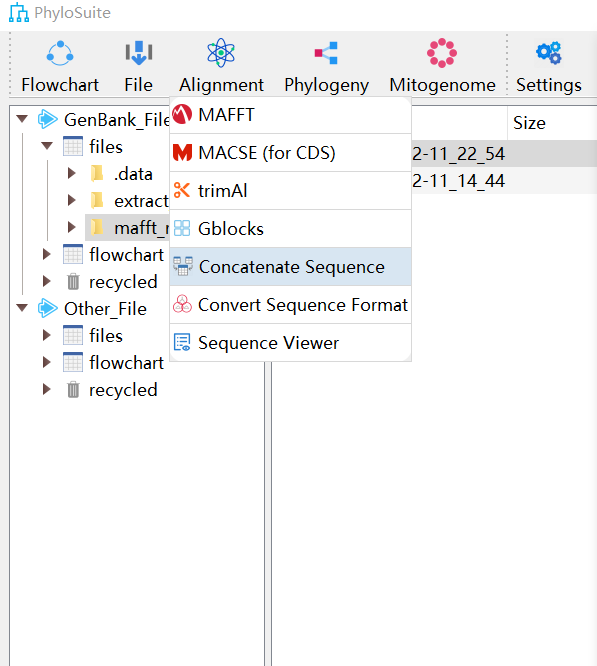
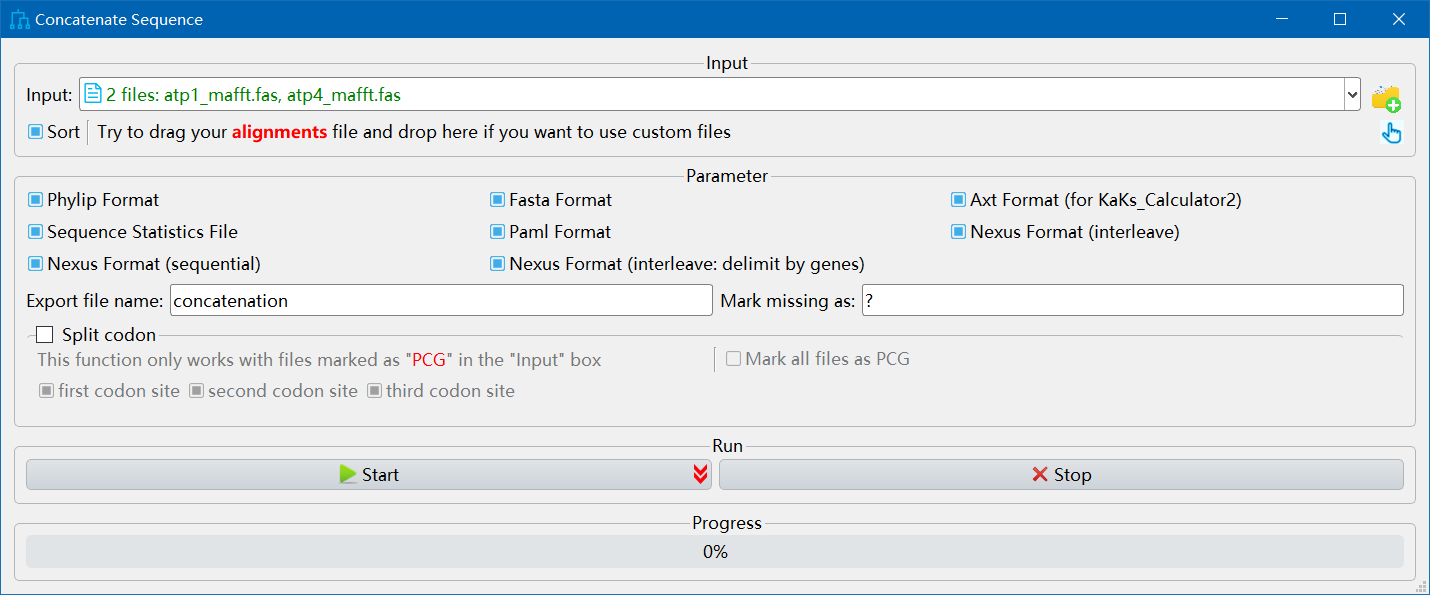
选择比对好的序列,进行连接
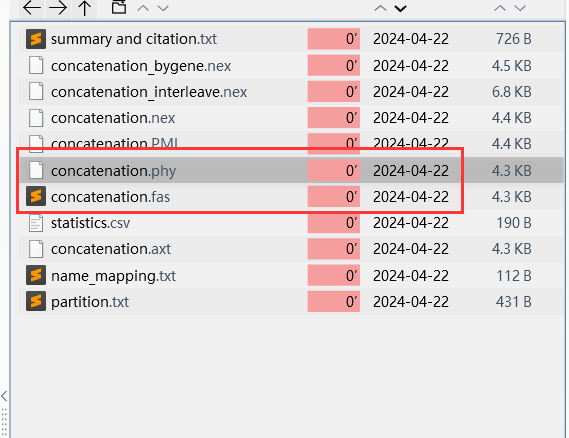
- IQ-Tree构建系统发育树
选择上面的.phy和.fas文件构建系统发育树,注意选择外群,其余参数可以不变
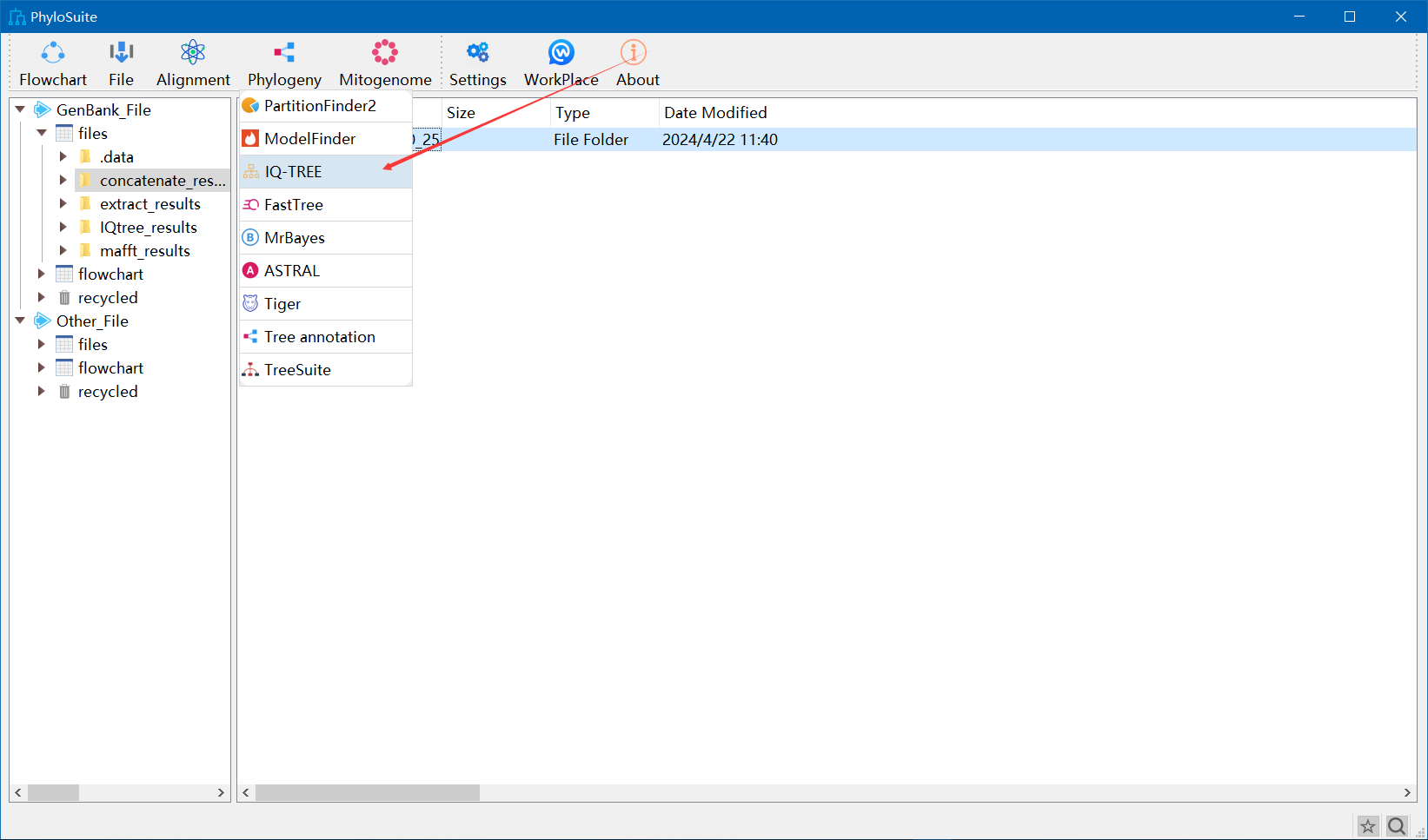
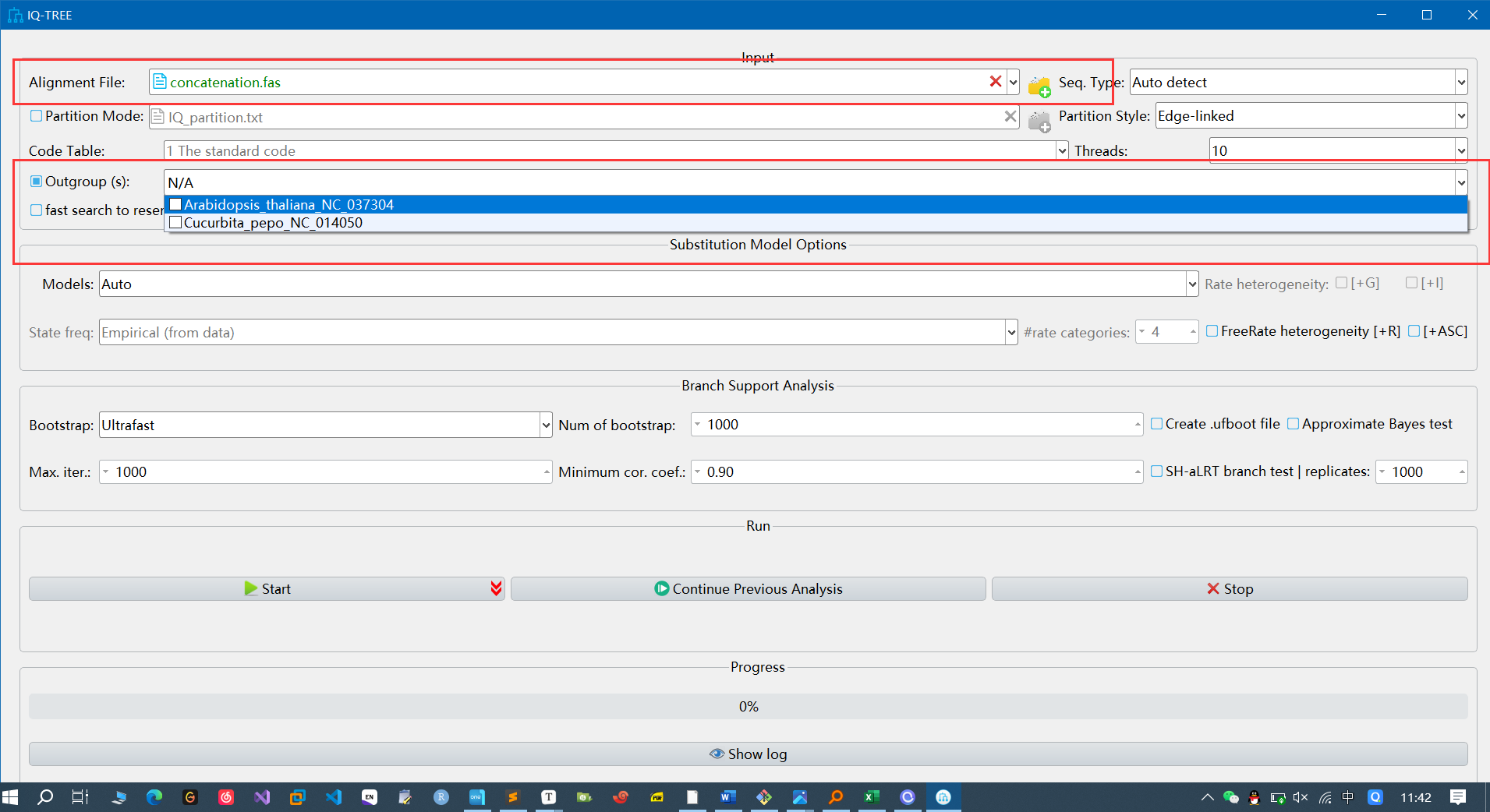
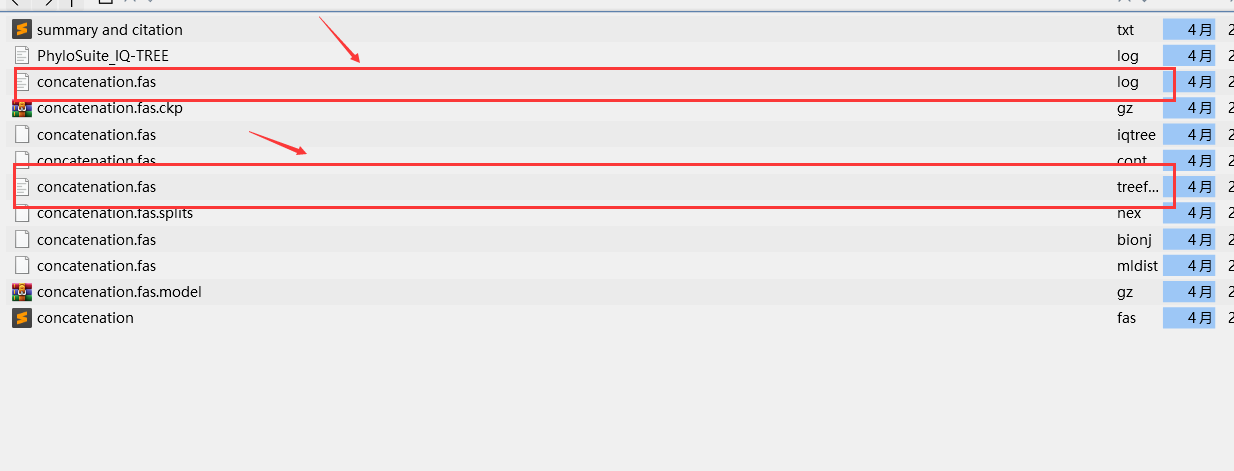
- 建树完成后,打开结果文件夹,iqtree选择的模型在.log文件夹中,可使用itol在线网站(iTOL: Interactive Tree Of Life (embl.de))对.treefile文件进行可视化和编辑
本博客所有文章除特别声明外,均采用 CC BY-NC-SA 4.0 许可协议。转载请注明来源 Sunning's Blog!
评论